
Invasive ductolobular carcinomas have a ductal origin
Treatment of non-small cell lung cancer (NSCLC), which represents 80-85% of lung cancer cases, has drastically changed over the past two decades with the development of molecularly targeted therapies, including tyrosine kinase inhibitors. Mutations in the epidermal growth factor receptor (EGFR) gene occur in over 30% of NSCLC cases, and EGFR tyrosine kinase inhibitors such as Osimertinib are frequently used as first-line therapies for EGFR-mutant NSCLC. However, most lung cancers acquire resistance to Osimertinib therapy within an average of 10 months, most commonly through secondary EGFR mutations, MET amplification, and histological transformation to small cell lung cancer. In some cases (approximately 10%), fusions involving oncogenes may play a role in Osimertinib resistance, and combination therapies targeting the mutant EGFR and the acquired oncogenic fusion partners may prove worthwhile. In this study, the authors evaluate 104 unique gene fusions as potential contributors to Osimertinib resistance in EGFR-mutant lung cancer. In addition, using an in vitro cell model, they test the ability of combination therapies to overcome Osimertinib resistance that results from gene fusions, providing insights for clinical practice. They also identify mechanisms by which cells may eventually develop resistance to these combination therapies.

Genetic data were collected from 3647 patients with NSCLC who had undergone DNA sequencing with the OncoPanel NGS assay at Dana-Farber Cancer Institute, Boston, MA, USA. Of these patients, 504 were found to have EGFR L858R or deletion in exon 19 (del19) mutations. After eliminating intragenic fusions and duplicates, 104 unique fusions from 85 patients with sensitizing EGFR mutations were identified. These fusions were classified into five groups; 37 fusions that included known oncogenes (16 lung cancer oncogenes and 21 oncogenes reported in other cancers) were selected for further study. To determine the clinical significance of these gene fusions, RNA sequencing followed by bioinformatics analyses were conducted on samples from 11 patients that had developed Osimertinib resistance. As opposed to DNA sequencing, which detects all gene fusions, RNA sequencing can identify which fusion events are expressed, allowing for interpretations of clinical significance. From the RNA sequencing data of this subset of patients, only one oncogenic fusion, DLG1-BRAF, was consistently detected by 3 different fusion callers. Even lowering the threshold to detection by 2 fusion callers failed to validate any other oncogenic fusion as expressed.
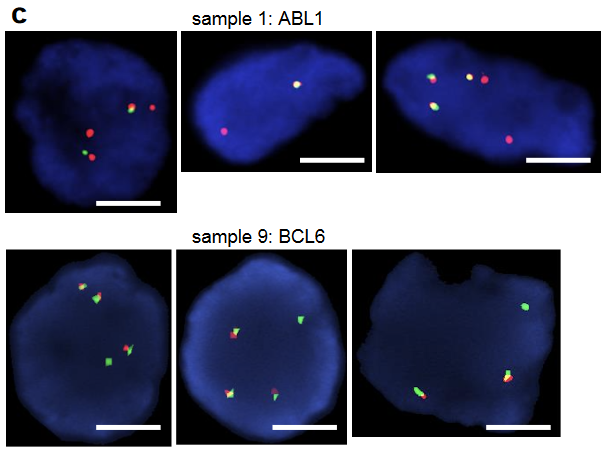
Orthogonal techniques were employed to confirm these RNA sequencing results, including both RT-PCR and FISH. The DLG1-BRAF fusion that had been detected by OncoPanel and RNA-seq was confirmed by RT-PCR and Sanger sequencing. A very faint band representing a FKBP5-ESR1 fusion was detected by RT-PCR. Sanger sequencing revealed that the fusion breakpoint was in the FKBP5 exon untranslated region after the stop codon; thus, the fusion protein would not be expressed. Similarly, structural variants in ABL1 or BCL6 were identified by FISH, but again, the data suggest that these did not produce functional fusion proteins; this confirmed that the RNA sequencing protocol accurately reflected expressed fusion proteins. Overall, although many fusions were identified by DNA-based sequencing, most of these did not lead to oncogenes capable of mediating drug resistance. This is clinically important, as it means that not all gene rearrangements are mechanisms of drug resistance, and therefore combination treatments aimed at many of these putative fusions may not elicit a therapeutic response. This was exemplified in one presented case. A patient with an EGFR del19 and T790M adenocarcinoma was treated with Osimertinib for 4 years, when a GKAP1-NTRK2 fusion was identified. Based on reports of GKAP1-NTRK2 in gliomas, this patient was treated with a combination of Osimertinib and Larotrectinib, but no therapeutic response was achieved. Further examination of the RNA-seq data revealed that the NTRK2 isoform did not have a kinase domain and so cannot be functional, resulting in the lack of response to Larotrectinib.
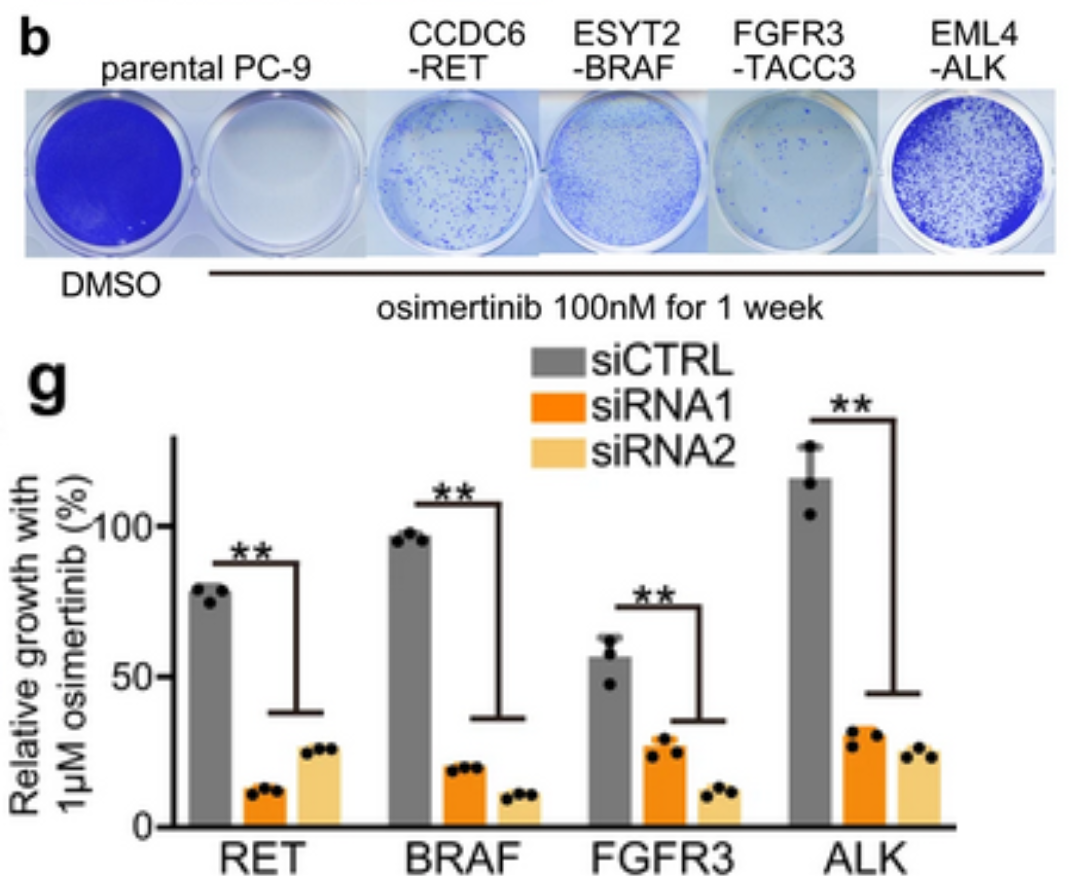
To further clarify which of the fusions identified by DNA sequencing were functional, an in vitro model system was created using CRISPR/Cas-9 to create fusion genes in the EGFR del19 mutant lung cancer cell line PC-9. Five lung cancer oncogene fusions that had been detected by OncoPanel NGS were selected to model in vitro: DLG1-BRAF, which had also been detected by RNA-seq, and CCDC6-RET, ESYT2-BRAF, FGFR3-TACC3, and EML4-ALK, which had been identified in a previous study as potentially contributing to TKI resistance. Edited PC-9 cells containing these fusion genes grew under Osimertinib treatment, demonstrating that these fusions do have a functional role in Osimertinib resistance. Furthermore, knockdown of RET, BRAF, FGFR3, or ALK genes in these CRISPR-edited cells resensitized them to Osimertinib treatment, corroborating the link to Osimertinib resistance. Combination treatments were tested in these resistant cells, and highly selective TKIs such as pralsetinib, selpercatinib, erdafitinib, and alectinib were most effective at inducing growth inhibition and apoptosis over time. In addition, chronic exposure of these cell lines to combination therapies revealed a wide variety of resistance mechanisms that developed, including YAP1 amplification as a mechanism of resistance to RET inhibitors. The authors propose that the AURK inhibitor alisertib could overcome resistance mediated by YAP1 amplification. More research is needed to build on these findings to ensure effective and targeted treatment for patients undergoing combination therapies for EGFR-mutant NSCLC.
