
Extrachromosomal MET Amplification Spurs Drug Resistance in ROS1+ NSCLC
Non-small-cell lung cancer (NSCLC) is a genetically heterogeneous disease. In approximately 2% of NSCLC cases, the primary oncogenic mutation is a chromosomal rearrangement involving ROS1 (ROS proto-oncogene 1), where one of many diverse gene partners fuses to the 3’ kinase domain of ROS1 and results in the constitutive activation of the ROS1 receptor tyrosine kinase. The ROS1-targeted tyrosine kinase inhibitor (TKI) entrectinib is an FDA-approved treatment for patients with these ROS1 fusions (ROS1+ NSCLC) and has dramatically improved patient outcomes. However, patients will eventually experience disease progression on entrectinib therapy as drug resistance develops. As a result, it is imperative to determine the underlying mechanisms of acquired resistance to determine the best sequential therapies. For example, next-generation ROS1-TKIs are being developed for use in refractory ROS1+ NSCLC patients with on-target mutations to the ROS1 kinase domain. However, these mutations have only been identified in approximately one-third of patients resistant to entrectinib; the mechanisms of resistance in most patients have yet to be elucidated. The aim of this study is to investigate these less understood mechanisms of entrectinib resistance in ROS1+ NSCLC.
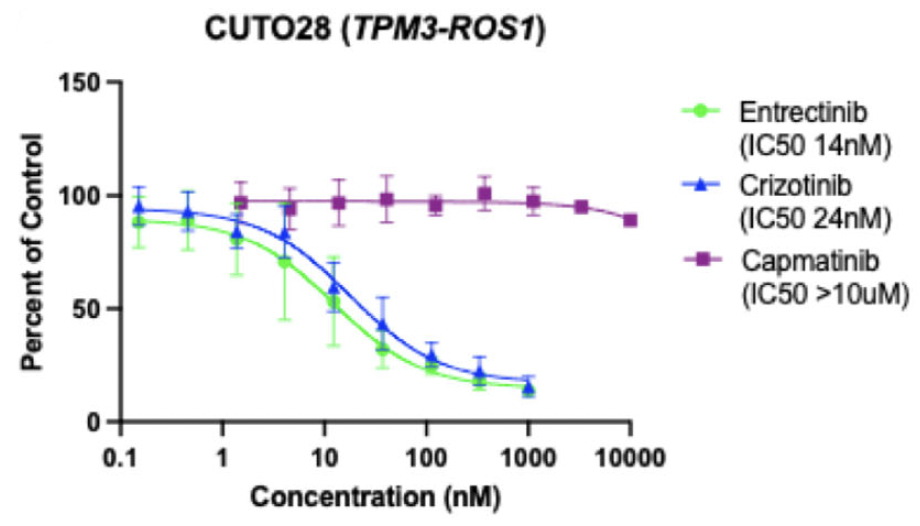
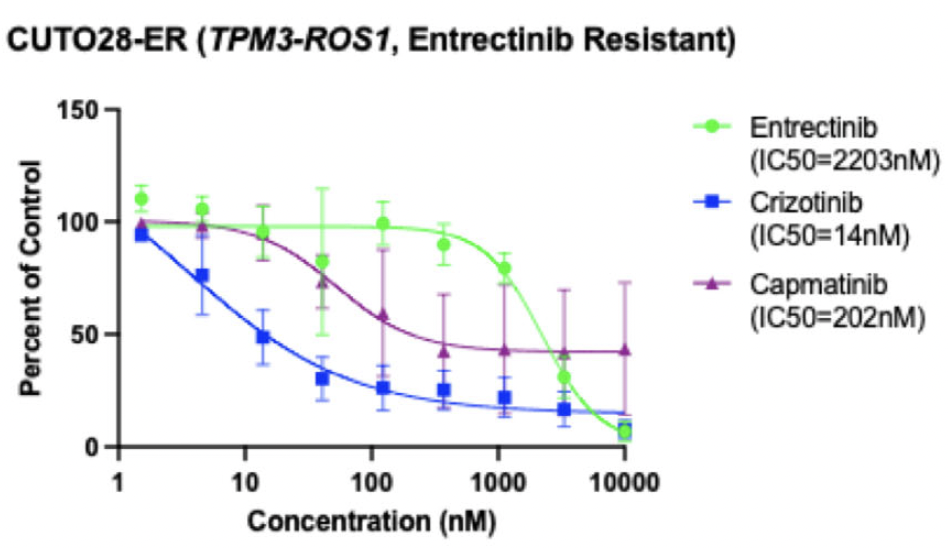
The CUTO28 cell line was generated from the pleural effusion of a patient with ROS1+ NSCLC. Genome alignment of RNA sequencing data confirmed a ROS1 fusion with TPM3 (Tropomyosin 3) as the 5’ gene partner. This cell line was initially sensitive to entrectinib, so to generate an entrectinib-resistant cell line (CUTO28-ER), CUTO28 cells were exposed to increasing doses of entrectinib until they were stably proliferating at a concentration of 500 nM (approximately 40x the IC50 of parental cells). Whole exome sequencing of the resulting CUTO28-ER cell line did not reveal a mutation in the ROS1 kinase domain; thus, an unknown resistance mechanism was present. The IC50 of the CUTO28-ER cells to entrectinib was over 2μM (157x the IC50 of parental cells). However, the CUTO28-ER cells were more sensitive to the ROS1/MET inhibitor crizotinib than the parental CUTO28 cells (IC50 of 14nM vs. 24 nM). This suggested that MET may be contributing to resistance in the CUTO28-ER cell line. To confirm, both CUTO28-ER and the parental CUTO28 cell lines were exposed to capmatinib, a MET-selective TKI that does not target ROS1. The parental cells were completely resistant (IC50 > 10μM) whereas the CUTO28-ER cell line was sensitive to capmatinib (IC50 = 202 nM), suggesting that these cells were more reliant on MET signaling for growth than the parental cells.
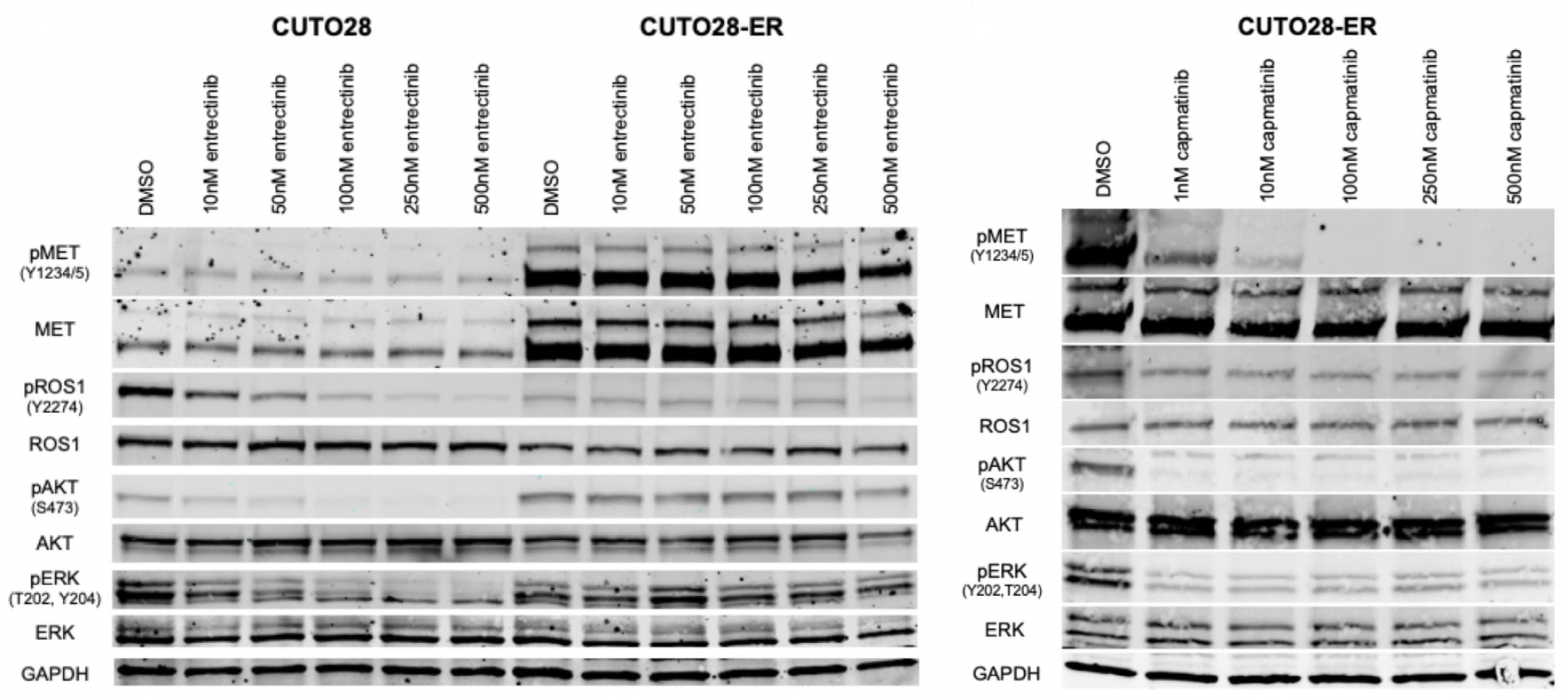
Immunoblotting was used to investigate whether CUTO28-ER cells were bypassing ROS1 signaling via activation of MET protein. To this end, western blot consistently showed low levels of phosphorylated ROS1 (pROS1) in CUTO28-ER cells, both in the absence and presence of entrectinib. Downstream targets of ROS1, including activated Akt (pAkt) and activated ERK1/2 (pERK1/2) were decreased by entrectinib in CUTO28 parental cells but were not affected by entrectinib in CUTO28-ER cells. Inhibition of MET using capmatinib did decrease this downstream signaling in CUTO28-ER cells. Both total MET and phosphorylated MET (pMET) were greatly increased in CUTO28-ER cells compared to the parental cell line. RNA sequencing showed a drastically significant increase in MET expression compared to the parental cell line (p = 2.78 x 10 -64). Collectively, these data suggest that CUTO28-ER cells are no longer dependent on ROS1 activation and instead are upregulating the production and activation of MET, leading to entrectinib resistance.

Next, fluorescent in situ hybridization (FISH) probes targeting MET and the chromosome 7 centromere (CEP7 control probe) were used to determine if CUTO28-ER cells had increased the expression and activation of MET through MET amplification. The parental CUTO28 cells had a MET:CEP7 ratio of 1.0, whereas the CUTO28-ER cells had a MET:CEP7 ratio of 4.2. This signified MET amplification. Metaphase FISH was used to determine if this observed amplification was occurring intrachromosomally and/or extrachromosomally. The mean number of extrachromosomal DNA (ecDNA) signals was 76.67 in the CUTO28-ER cells and 0.00 in parental CUTO28 cells. This showed that the amplification was occurring exclusively outside of the genomic DNA through ecDNA. Genomic qPCR found the ecDNA expression of MET to be 7.84x greater that of genomic DNA in CUTO28-ER cells. The authors found no evidence of other known mechanisms of increasing MET activation (e.g., MET exon 14 skipping, Y1003 mutation, activating point mutations in the MET kinase domain, MET fusions). Thus, the data suggests that ecDNA MET gene amplification was responsible for MET protein activation and led to bypass signaling in this entrectinib-resistant NSCLC cell line. To determine the clinical relevance of this discovery, the authors examined circulating tumor DNA (ctDNA) of 105 ROS1+ patients enrolled in the STARTRK-2 phase 2 basket study of entrectinib. Two of these patients (1.9%) were found to have MET copy number alterations; however, as the successful detection rate of ROS1 fusions via ctDNA in this trial was only 65% and copy number variations are more difficult to detect in plasma than ROS1 fusions, it is possible that the rate of MET amplification in ROS1+ NSCLC is even greater than this. This study highlights the need for clinical monitoring for MET amplifications in refractory ROS1+ NSCLC patients, as MET-mediated bypass signaling can be inhibited with crizotinib, capmatinib, or tepotinib to improve patient outcomes.

Learn more about our MET probe
https://empiregenomics.com/product/16266Citation & Source Link
Tyler LC, Le AT, Chen N, Nijmeh H, Bao L, Wilson TR, Chen D, Simmons B, Turner KM, Perusse D, Kasibhatla S, Christiansen J, Dudek AZ, Doebele RC. MET gene amplification is a mechanism of resistance to entrectinib in ROS1+ NSCLC. Thorac Cancer. 2022 Nov;13(21):3032-3041. doi: 10.1111/1759-7714.14656. Epub 2022 Sep 13. PMID: 36101520; PMCID: PMC9626307.
https://onlinelibrary.wiley.com/doi/10.1111/1759-7714.14656