
Loss of TP53 Promotes NEK2 Amplification, Spurring Multiple Myeloma Aggressiveness
In the broadest sense, cancer development is spurred by genetic abnormalities that result in oncogene activation and tumor suppressor gene inactivation. In multiple myeloma (MM), a hematological malignancy that affects plasma cells, the amplification of chromosome 1q and the deletion of chromosome 17p have both been shown to be related to disease progression and poor patient outcomes. Chromosome 1q contains several oncogenes including NEK2 (never in mitosis gene A-related kinase 2), which has been shown to promote tumor cell proliferation, metastasis, and drug resistance in MM and other cancer types. Chromosome 17p contains the tumor suppressor gene TP53 (tumor protein p53), which is an independent prognostic factor in MM and is known to be mutated or deleted in many cancer types. In this study, the authors investigate the relationship between aberrant NEK2 and TP53 expression. Previous research has identified a non-canonical p53 binding site within the distal promoter region of NEK2; as a result, the authors hypothesize that the combined loss of TP53 and amplification of NEK2 could produce a synergistic effect that results in especially poor outcomes for MM patients with both defects.
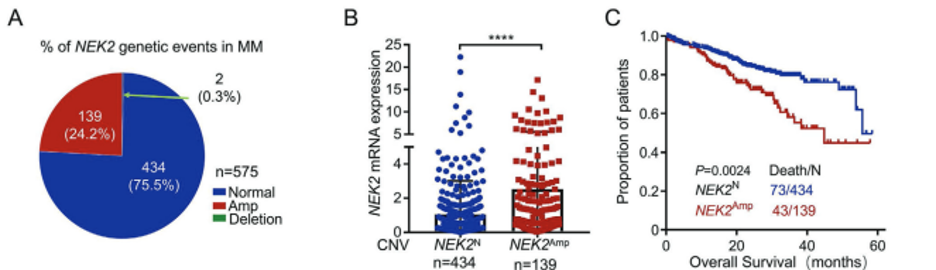
The Multiple Myeloma Research Foundation CoMMpass database was used to examine the connection between genetic abnormalities, NEK2 expression, and patient outcomes. Using DNA copy number variation data from 575 samples, the authors determined that 24.2% (139/575) of MM patients had NEK2 amplification. Amplification showed a significant correlation with NEK2 mRNA overexpression (p < 0.0001) in these samples. Patients with NEK2 amplification had a median overall survival of 40 months, which was significantly shorter than the median overall survival of patients with a normal NEK2 copy number (60 months). Using the same database, the authors next assessed the correlation between NEK2 and TP53 abnormalities. TP53 deletion occurred in 9.3% of 548 MM samples and was correlated with NEK2 overexpression (p < 0.0001) and an increased proportion of patients with NEK2 amplification. Patients with both NEK2 amplification and TP53 deletion had the worst overall survival, at 32.4 months, compared to those with only TP53 deletion (49 months), only NEK2 amplification (44.8 months), or neither NEK2 amplification nor TP53 deletion (not reached). To confirm the correlations detected in this database, human bone marrow samples were collected from 8 healthy donors, 82 newly diagnosed and 25 relapsed MM patients at the Third Xiangya Hospital and Peking Union Medical College for analysis. Fluorescent in situ hybridization (FISH) demonstrated that 23.5% (5/17 samples used for FISH) of newly diagnosed patients had NEK2 amplification, which recapitulated the CoMMpass database results. NEK2 amplification was detected by FISH in 100% (4/4) of samples with TP53 deletion but was not detected (0%) in 7 samples with wild-type TP53. Additionally, immunofluorescence conducted on samples from 51 newly diagnosed and 16 relapsed MM patients revealed that NEK2 protein expression is inversely correlated with p53 expression. Collectively, these results support a relationship between TP53 deletion and NEK2 upregulation, which contribute to a poor prognosis in MM.
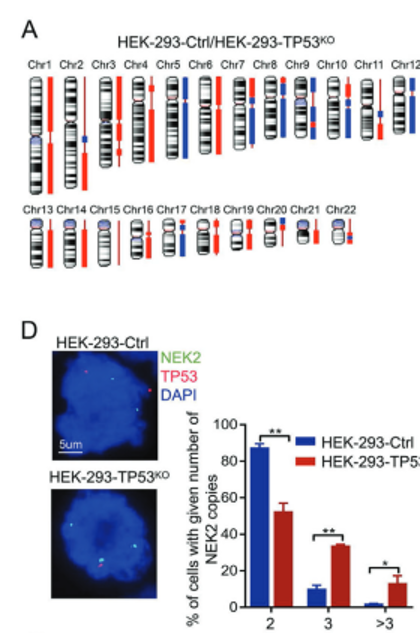
To establish a causal link between TP53 deletion and NEK2 overactivation, p53 expression was induced using different concentrations of nutlin-3a in TP53 WT and TP53-/- MM cell lines. Nutlin-3a inhibits MDM2-p53 interactions, stabilizing and activating the wild-type p53 protein. As nutlin-3a concentrations increased, TP53 mRNA and protein levels increased (p < 0.01) and NEK2 mRNA and protein levels decreased (p < 0.01), but only in the TP53 WT cell lines; without a wild-type copy of TP53, there was no functional p53 protein to be stabilized by nutlin-3a, so there was no effect of increasing nutlin-3a concentrations on NEK2 protein and mRNA levels. This suggests that WT p53 activity directly impacts NEK2 expression, and hints at a causal relationship. Knocking out TP53 using CRISPR-Cas9 editing triggered a dramatic increase in NEK2 mRNA and protein levels, and subsequent restoration of WT TP53 expression resulted in a decrease in NEK2 mRNA and protein levels. Thus, the data suggest that NEK2 expression is inversely related to TP53 activation in a cause-and-effect manner. In addition, using a comparative genomic hybridization (CGH) array, the authors determined that TP53-knockout HEK293 cells were especially prone to chromosomal instability, including deletion of chromosomes 5, 7q, 8, 9, and 17p and gains of chromosomes 1, 2q, 3p, 4p, 6, 13q, and 14q. Of note, chromosome 1q21-44, where NEK2 is located, was dramatically amplified; FISH confirmed that the number of cells with 3 and >3 copies of NEK2 significantly increased in the TP53 knockout cells (p < 0.01 and p < 0.05, respectively). Taken together, the data suggest that loss of TP53 leads to NEK2 amplification and overexpression in MM.
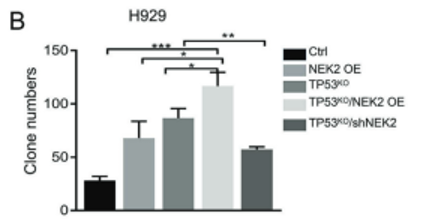
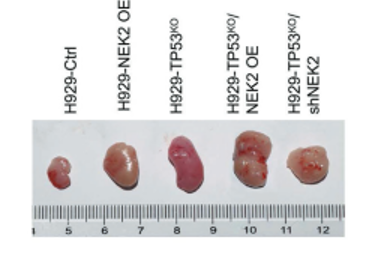
The final task was to determine if dual defects in TP53 and NEK2 result in a synergistic effect promoting MM development. To this end, NEK2 was overexpressed in H929-TP53 WT cells and in H929-TP53KO cells. Cells with dual defects in NEK2 and TP53 were most proliferative as determined by both clonogenic soft agar assay and BrdU intake assay. When these cell lines were injected subcutaneously into the abdomen of mice, the most dramatic tumor growth was seen in the dual defect group. As the authors previously found NEK2 overexpression to contribute to drug resistance in MM patients, they used an apoptosis assay to test the sensitivity of cell lines with dual defects to Bortezomib (BTZ), a first-line MM treatment. The percentage of apoptotic cells was lowest in the dual defects group (67.2%) after BTZ treatment when compared to control (96%). It was possible to reverse the proliferative, tumorigenic, and drug-resistant capacity of these cells with expression of wild type p53 in MM cell lines, and this reversal was even more substantial when combined with NEK2 depletion. These results show that dual defects in NEK2 and TP53 can have a significant clinical impact on MM patients and suggest the possibility of developing selective NEK2 inhibitors to improve therapeutic outcomes.

Learn more about our NEK2 probe
https://empiregenomics.com/fish-probes/gene/NEK2Citation & Source Link
Feng X, Guo J, An G, Wu Y, Liu Z, Meng B, He N, Zhao X, Chen S, Zhu Y, Xia J, Li X, Yu Z, Li R, Ren G, Chen J, Wu M, He Y, Qiu L, Zhou J, Zhou W. Genetic Aberrations and Interaction of NEK2 and TP53 Accelerate Aggressiveness of Multiple Myeloma. Adv Sci (Weinh). 2022 Mar;9(9):e2104491. doi: 10.1002/advs.202104491. Epub 2022 Jan 27. PMID: 35088582; PMCID: PMC8948659.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8948659/