PDGFRA mutation increases cell proliferation in vitro via an oncogenic splice variant
The era of personalized medicine and genome-based cancer therapies has been accelerated by next-generation sequencing technologies, which allow for the identification of gene variants with clinical implications for diagnosis, prognosis, and patient care. However, many newly identified variants have yet to be linked to disease, and these are referred to as variants of uncertain significance (VUS). In glioblastoma, an aggressive and treatment-resistant cancer of the central nervous system, a VUS of the PDGFRA gene, which encodes the platelet-derived growth factor receptor-α, has been identified in a subgroup of patients. As the availability of molecularly targeted drugs for brain tumors is limited, it is important to investigate this VUS for pathogenic significance and as a potential target for new therapies. In this study, the authors used CRISPR/Cas9 to generate a PDGFRA-mutated cell line that allowed them to investigate the role of the VUS in cell proliferation and assess the sensitivity of the mutant cell line to molecularly targeted drugs. They also studied the results of the mutation in three patients with glioblastoma.
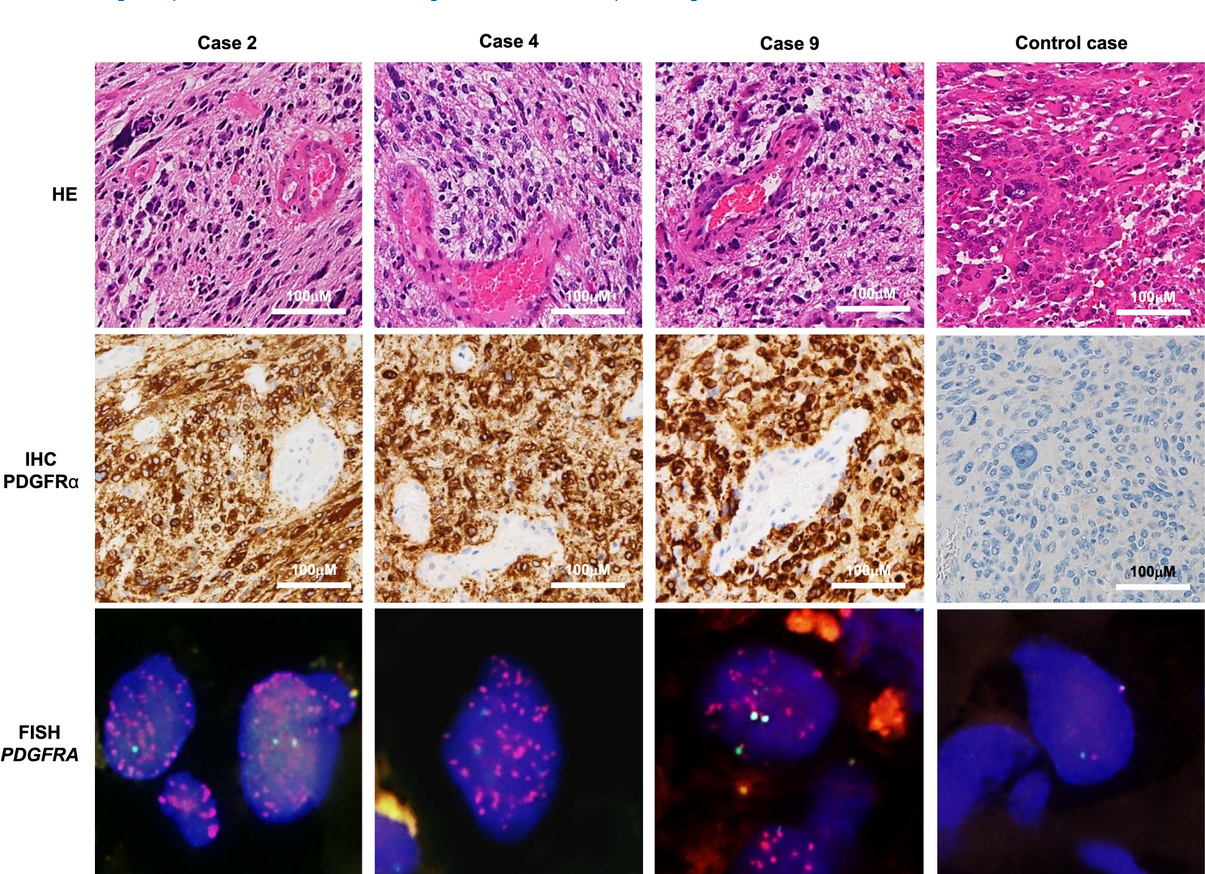
In a previous study, the authors had developed a glioma-tailored next-generation sequencing panel for molecular characterization of central nervous system tumors. The panel was designed to identify common genetic mutations in gliomas, including mutations in the isocitrate dehydrogenase 1 or 2 (IDH1 or IDH2) gene, codeletion of chromosomal arms 1p and 19q (1p19q codeletion), the H3‐K27M mutation, and others. This study identified a subgroup of glioblastoma patients (11/64; 17%) with wild-type IDH1, no 1p19q codeletion, and no TERT promoter mutation but that harbored mutations in PDGFRA. Most of the PDGFRA mutations were VUS or not registered in the JAX and OncoKB databases. For this study, the authors selected a VUS that was seen in three of these patients for further investigation (c.1403A > G in exon 10 of the PDGFRA gene). In addition to harboring this mutation, fluorescent in situ hybridization (FISH) revealed that these cases also exhibited 30- to 84-fold gene amplification of PDGFRA.

To study the role of the VUS in vitro, the authors used the microhomology-assisted excision (MhAX) method to generate human embryonal kidney 293 T (HEK293T) cells that contained the PDGFRA c. 1403A > G mutation and a silent mutation (“mutated”), along with a HEK293T cell line that only contained the silent mutation (“control”). PDGFRA mRNA expression was then examined in these cell lines using paired primers designed to include the mutation site. The PCR product from the mutated cell line was shorter than that of both the control cell line and parental HEK293T cell line, and Sanger sequencing revealed that the first 39 bp of exon 10 had been deleted in the mutated clone. This suggested that the c. 1403A > G mutation created a new 3’ splice site, resulting in intron elongation and a new PDGFRA splice variant. This in-frame PDGFRA splice variant was also found in 2/3 patients with the c.1403A > G mutation. The authors note that this rare splicing variant has also been identified in pediatric high-grade glioma cases. Thus, PDGFRA c.1403A > G is likely an oncogenic or pathogenic mutation.
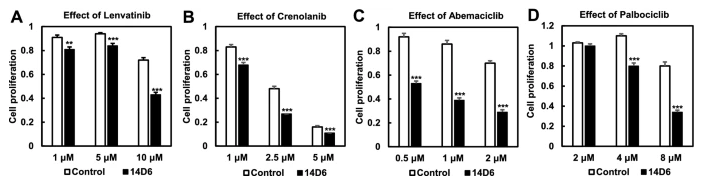
Next, to further investigate the oncogenic potential of the PDGFRA c.1403A > G mutation, the authors used RT-PCR to quantify the mRNA expression in both the mutated and control cell lines. The mutated clone had significantly higher PDGFRA mRNA expression than the control (p <0.001). Western blot analysis revealed that protein expression of PDGFRα did not increase as much as PDGFRA mRNA expression, and that protein levels were similar between control and mutated cell lines. However, there was an increase in the expression of tyrosine-phosphorylated PDGFRα in the mutated cells, suggesting some potential for this mutation to contribute to tumor proliferation. Indeed, the downstream components of the PDGFR signal transduction pathway showed increased phosphorylation, and expression of cyclinD1, CDK4, CDK6, and c-Myc significantly increased in the mutated cell line (p < 0.05 for cyclinD1, CDK4, and c-MYC and p < 0.01 for CDK6). In addition, the mutated cells had higher proliferative activity, as revealed by WST assay and trypan blue exclusion test, and showed increased cell numbers in the S and G2/M phase and decreased cell numbers in the G0/G1 phase compared to the control. Thus, it appears that cells with the c. 1403A > G mutation undergo a faster G1/S transition through PDGFRα-mediated activation of the CDK4/ CDK6-cyclin D1 pathway. Kinase inhibitor drugs targeting PDGFRα and the CDK4/CDK6 signaling pathway, including lenvatinib, crenolanib, abemaciclib and palbociclib, significantly reduced proliferation in the mutated cells in a dose-dependent manner. As there is a great need for better therapeutic interventions in glioblastoma, future studies should continue to investigate the role of this potentially oncogenic variant in the disease and assess the use of kinase inhibitors for treatment of patients harboring this variant.
