
Loss of chromosome Y linked to AML and clonal hematopoiesis
Mosaic loss of chromosome Y (mLOY) is the most common form of clonal mosaicism and is recurrent in the blood cells of adult males. This chromosome alteration typically increases with age and has been linked with aging-related diseases including clonal hematopoiesis (CH), in which a single hematopoietic stem and progenitor cell (HSPC) gives rise to a genetically distinct subpopulation of peripheral blood or bone marrow cells, and acute myeloid leukemia (AML). However, functional evidence of a causal relationship between mLOY and the development of these diseases is still lacking, due in part to the technical challenges of precisely deleting a whole chromosome in somatic cells. Previous studies using XO mice have failed to find a link between mLOY and leukemogenesis, though this could be due to the differing DNA damage repair mechanisms in embryonic stem cells and somatic cells. Thus, the authors hypothesized that mLOY in somatic cells, but not XO in the germline, might drive malignant transformation. To test this hypothesis, they used a recently described CRISPR/Cas9-mediated targeted chromosome elimination technique to generate mLOY in mouse HSPCs, then examined the effects of mLOY on the development of CH and AML.
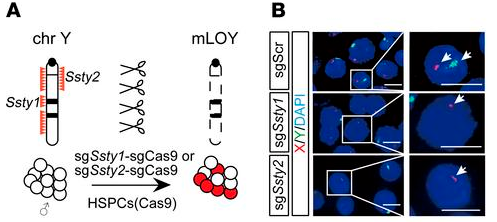
First, two single guide RNAs (sgRNAs) were designed to target Ssty1 (spermiogenesis-specific transcript on Y 1) and Ssty2 (spermiogenesis-specific transcript on Y 2) on mouse chromosome Y. These frequently repeated sequences are unique to the Y chromosome, and targeted deletion of these regions has been shown to result in the loss of chromosome Y with minimal off-target effects. The Ssty sgRNAs were independently introduced into c-Kit+ HSPCs from Rosa-Cas9 mice; control (sgSCR-sgCas9) HSPCs were generated by targeting a non-gene region on mouse chromosome 8. Fluorescence in situ hybridization (FISH) was used to visualize the resulting mLOY, which was seen in approximately 15% of sgSsty1-sgCas9 and sgSsty2-sgCas9 HSPCs and not at all in the control sgSCR-sgCas9 cells. The edited HSPCs were then transplanted into sublethally irradiated mice. FISH analysis of bone marrow samples at 12 and 21 weeks after transplantation revealed a marked increase in mLOY in sgSsty1-sgCas9 and sgSsty2-sgCas9 mice, from approximately 15% in preinjected cells to around 30% at 12 weeks after transplantation (p < 0.05) and more than 50% at 21 weeks after transplantation (p < 0.01). This increase in proportion of cells with mLOY is like the clonal expansion seen in CH and suggested the possibility that mLOY cells have a competitive growth advantage. There were also small but significant reductions in RBC counts in the peripheral blood of sgSsty1-sgCas9 and sgSsty2-sgCas9 mice compared to the control mice, which resembled the anemia that frequently accompanies mLOY in elderly male patients with CH.

Next, the authors investigated whether mLOY could lead to DNA damage, increasing the likelihood of malignant transformation to AML. Immunofluorescence results indicated that both sgSsty1-sgCas9 and sgSsty2-sgCas9 HSPCs had significantly increased DNA damage compared to control cells, as measured by the comet assay and γH2AX staining. Further, when female (XX) HSPCs were edited with sgSsty1 and sgSsty2, they did not experience an increase in DNA damage compared to the control cells. Thus, the data suggest that it is the loss of the Y chromosome that leads to increased DNA damage in HSPCs, possibly contributing to malignant transformation in AML. To test this idea, the authors transduced sgSsty1-sgCas9, sgSsty2-sgCas9, or sgSCR-sgCas9, along with AML1-ETO into Trp -/-; Cas9 HSPCs. An analysis conducted by the authors of 3 independent AML patient cohorts showed that mLOY was significantly associated with AML1-ETO fusion and TP53 deficiency, which informed the design of their model HSPC system. The edited HSPCs were transplanted into sublethally irradiated wild-type recipient mice. After 7 weeks, mice that received sgSsty1 or sgSsty2 in addition to AML1-ETO were more likely to have WBC counts above 40 x 109/L than those that received AML1-ETO alone, though this result was not significant. In addition, the cells with mLOY and AML1-ETO had a significantly shorter latency to developing AML (56 days and 56 days vs. 64 days), suggesting that mLOY may accelerate leukemogenesis. FISH analysis of the resulting AML cells also showed an expansion of mLOY in the sgSsty1 and sgSsty2 cells (p < 0.05 and p < 0.01, respectively).
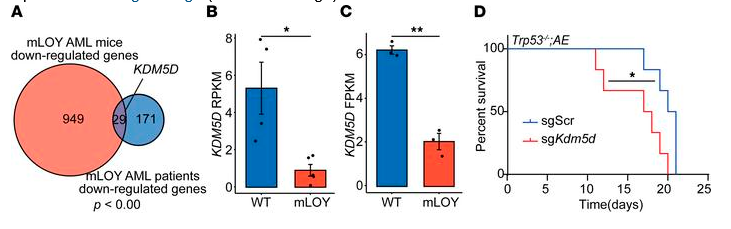
Finally, the authors conducted an examination of commonly downregulated genes in both human and mouse AML with mLOY and identified the loss of lysine demethylase 5D (KDM5D), a chromosome Y-specific H3K4 demethylase, as a potential driver mutation. KDM5D has been suggested as a tumor suppressor in prostate cancer, and low KDM5D expression has been reported in AML with AML1-ETO translocation, but its specific role in AML is still unknown. So, to test its function in AML, the authors used CRISPR/Cas9 to disrupt Kdm5d in their murine HSPC line. The sgKdm5d HSPCs had significantly more DNA damage, as measured by the comet assay and γH2AX staining. Mice that developed AML after receiving Kdm5d-edited HSPCs survived an average of 17 days, compared to an average survival of 20 days in sgSCR AML mice (p < 0.05). Analysis of RNA-seq data from both mouse and human mLOY AML revealed that the genes significantly upregulated in the sgKdm5d HSPCs also showed greater expression in both AML patients and mice with mLOY; genes that were significantly downregulated in the sgKdm5d HSPCs showed less expression in AML patients and mice with mLOY. Thus, Kdm5d loss recapitulated mLOY in murine HSPCS, and the authors conclude that mLOY promotes AML and CH partially through the loss of Kdm5d. However, given the complex nature of the cell populations involved in AML and CH, future studies are needed to further assess the effects of mLOY and KDM5D loss in the development of these diseases.
