
Dusp4 needs help to trigger breast cancer tumorigenesis
The aggressive basal-like breast cancer (BLBC) subtype, which strongly correlates with triple-negative (ER- PR- HER2-) breast cancers, comprises 15-20% of all breast cancer cases and has a poor prognosis, largely resulting from a lack of a molecularly-targeted treatment regimen. Loss of control of signaling pathways frequently triggers tumorigenesis, and molecules involved in these signaling pathways are frequent molecular treatment targets. However, the mechanisms of tumorigenesis in BLBCs have yet to be fully elucidated. Dual-specificity phosphatase-4 (DUSP4) is a tumor suppressor gene that is frequently lost in aggressive cancer subtypes, including BLBC. DUSP4 encodes a suppressor of the mitogen-activated protein kinase (MAPK) pathway, which is intricately involved in the regulation of the cell cycle, and loss of DUSP4 can result in unrestricted MAPK signaling and cell cycle checkpoint escape. Low DUSP4 gene expression in BLBCs has been associated with increased tumor cell proliferation, poor prognosis, cancer stem cell-like functionality and chemotherapy resistance. However, targeted deletion of Dusp4 in murine models has not been shown to directly trigger tumorigenesis. As there are other frequent alterations in BLBCs, such as inactivating mutations in TP53 (p53) and amplification of MYC, it is possible that a combination of oncogenic events is required to trigger tumor growth in BLBCs. Here, the authors test whether the combination of Dusp4 loss with these additional oncogenic events can promote tumorigenesis in mouse breast cancer models.

Cre-Lox recombination was used to generate floxed Dusp4 mice, which were then crossed with MMTV-Neu-Ires-Cre (MMTV-NIC) mice to generate a line in which Dusp4 was deleted from Neu+ tumor cells. A decrease in Dusp4 mRNA was noted in the resulting tumors, and biallelic deletion of Dusp4 resulted in modest upregulation of the known Dusp4 phosphatase targets ERK1/2 and JNK; but, Dusp4 loss alone did not result in mammary gland hyperplasia, and biallelic Dusp4 deletion actually prolonged tumor latency. Thus, as reported in earlier studies, Dusp4 loss alone was not enough to trigger tumorigenesis. However, the MMTV-NIC model is more luminal-like in nature and does not replicate events typically incurred in the development of BLBCs, like TP53 loss and MYC amplification. So, to create a model more similar to BLBCs, primary mammary epithelial cells (PMECs) from a floxed Dusp4 mouse were used to generate a population of progenitor-like epithelial cells; these were transiently infected with either Ad-GFP to generate a Dusp4-competent control cell line, or Ad-Cre-mCherry to generate a Dusp4-deficient cell line. CRISPR/Cas-9 was then used to generate derivative cell lines with either wildtype p53 or edited Trp53 (p53 loss of function). Each of these lines were transduced with human cMYC (MYCAMP) or LACZ (MYCWT). Thus, their PMEC model could be used to test whether loss of Dusp4, in combination with p53 loss and/or MYC amplification, resulted in mammary gland tumorigenesis.
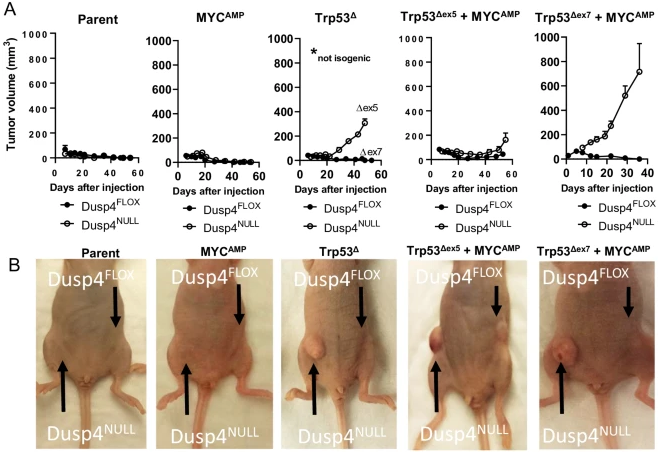
The derivative cell lines were orthotopically implanted in opposing #5 mammary fat pads of athymic nu/nu mice. The combined loss of Dusp4 and Trp53 led to the largest increase in tumor volume, and this trend was observed regardless of MYC amplification, although the most aggressive cell line also harbored MYC amplification. Though the most robust tumorigenic response was seen from Dusp4-deficient lines, one cell line with competent Dusp4 produced tumors; this cell line had p53 loss of function and MYC amplification, and the tumors produced were smaller and less aggressive than cell lines that were Dusp4-deficient. However, further investigations also suggested that Dusp4 loss was not directly impacting tumorigenesis. For example, when Dusp4 was abruptly deleted from Dusp4-competant Trp53∆ + MYCAMP cells just one week prior to injection into the host mice, no tumor growth was observed. Similarly, lentiviral reconstitution of human DUSP4 into Dusp4-deficient Trp53∆+ MYC AMP cells did not suppress tumor formation. Thus, the data suggest that the tumor-forming potential was not resulting from canonical Dusp4 function in MAPK signaling. Rather, as Dusp4 is also involved in G1/S checkpoint control, Dusp4 loss accompanied by loss of p53 function could result in genomic instability that helps to generate or select for the emergence of tumorigenic clones. Indeed, Dusp4 loss was associated with multinucleation in their cell lines, which the authors infer could be due to replication stress, and which could lead to genomic instability in the form of chromosome mis-segregation and copy number variations.
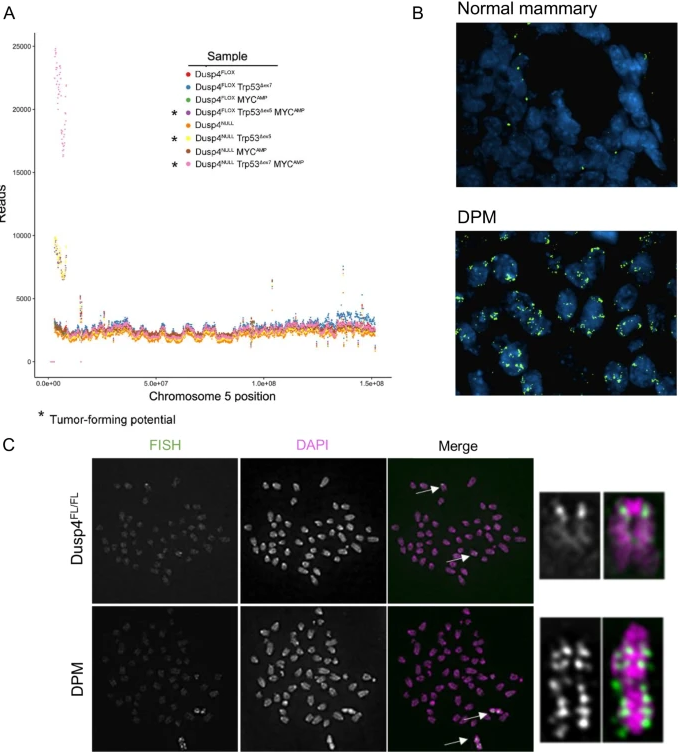
Whole-genome sequencing revealed an amplicon on murine chromosome 5 in all Dusp4-deficient lines, as well as in the Dusp4-competent Trp53 ∆+ MYCAMP cell line that formed small, slow-growing tumors. Because the amplicon was found in all tumor-forming lines, including one Dusp4-competent line, the authors hypothesize that Dusp4 loss is not required to generate this amplicon, but may select for it or facilitate its formation. The amplicon was located near the chromosome 5 centromere and contained the genes Dbf4, Cdk6, and Cdk14, which are all associated with cell cycle regulation. Fluorescent in situ hybridization confirmed amplification of this region in the Dusp4-deficient Trp53∆+ MYCAMP cell line, and metaphase spreads showed tandem duplications of the region throughout chromosome 5, suggesting repeated chromosome mis-segregation. When Dbf4 was silenced in the Dusp4-deficient Trp53∆+ MYCAMP cell line, and the resulting cells were orthotopically implanted into athymic nu/nu mice, the tumor-forming capacity of these cells, which had been aggressive in earlier trials, was greatly suppressed. Thus, one possible mechanism of BLBC tumorigenesis is emerging; loss of Dusp4, combined with loss of p53 function and potentiated by MYC overexpression, results in cells that acquire Dbf4 amplification through genomic instability and selective pressure; the action of Dbf4, which can rescue stalled DNA replication forks and initiate DNA replication, allows cells to escape cell cycle checkpoints and triggers tumorigenesis. Further studies will be needed to confirm and expound upon this proposed novel oncogenic mechanism in BLBCs.
