
CLIP1-LTK Fusion Identified as New Druggable Target in Lung Cancer
Non-small cell lung cancer (NSCLC) is the most common form of lung cancer and a leading cause of death worldwide. Fortunately, the survival rate for NSCLC patients has been increasing, in large part due to advances in targeted molecular and immunotherapies. Somatic mutations and gene fusions that alter the receptor tyrosine kinase (RTK)/RAS/REF pathway are frequent treatment targets; these druggable oncogenic alterations include KRAS, EGFR, and BRAF mutations as well as ROS1, ALK, and RET fusions. However, 25-40% of NSCLC cases lack known actionable oncogenic mutations. In this study, the authors describe a novel oncogenic fusion of CLIP1 (CAP-Gly Domain Containing Linker Protein 1), located on 12q24, with LTK (Leukocyte receptor Tyrosine Kinase), located on 15q15. CLIP1 is a known fusion partner of other receptor tyrosine kinases, including ROS1, ALK, and RET. The LTK protein is grouped in the same receptor tyrosine kinase subfamily as ALK and can activate the RAS/MAPK and PI3K/AKT pathways, but had not been definitively linked to oncogenesis. Here, the authors present the first evidence of a functional oncogenic alteration in LTK in cancer. Importantly, they also demonstrate that the CLIP1-LTK fusion is an actionable drug target for NSCLC patients that lack other known lung cancer biomarkers.

The LC-SCRUM-Asia cohort, which has enrolled more than 13,000 lung cancer patients from 307 institutes across Japan, was used to search for novel oncogenic drivers in patients that lack previously described mutations such as EGFR, ALK, ROS1, BRAF, MET, NTRK, HER2, RET, NRG1, and KRAS. Whole transcriptome sequencing of 75 consecutive samples analyzed between October 2020-December 2020 resulted in the identification of an in-frame CLIP1-LTK fusion in one patient. Reverse transcription-polymerase chain reaction (RT-PCR) confirmed the presence of these fusion transcripts in tumors from two metastatic sites. Fluorescent in situ hybridization (FISH) using a break-apart LTK probe showed solitary green signals (representing LTK rearrangement) in 82% of scored tumor cells, compared with the normal fused orange-green signals seen in non-tumoral cells. Both these tests confirm the presence of CLIP-LTK fusion in tumor cells. Sanger sequencing showed that the fusion occurred between exon 16 of CLIP1 and exon 11 of LTK in the expressed transcript. This fusion would place multiple coiled-coil domains found in CLIP1 upstream of the full LTK kinase domain and could result in constitutive kinase activation.
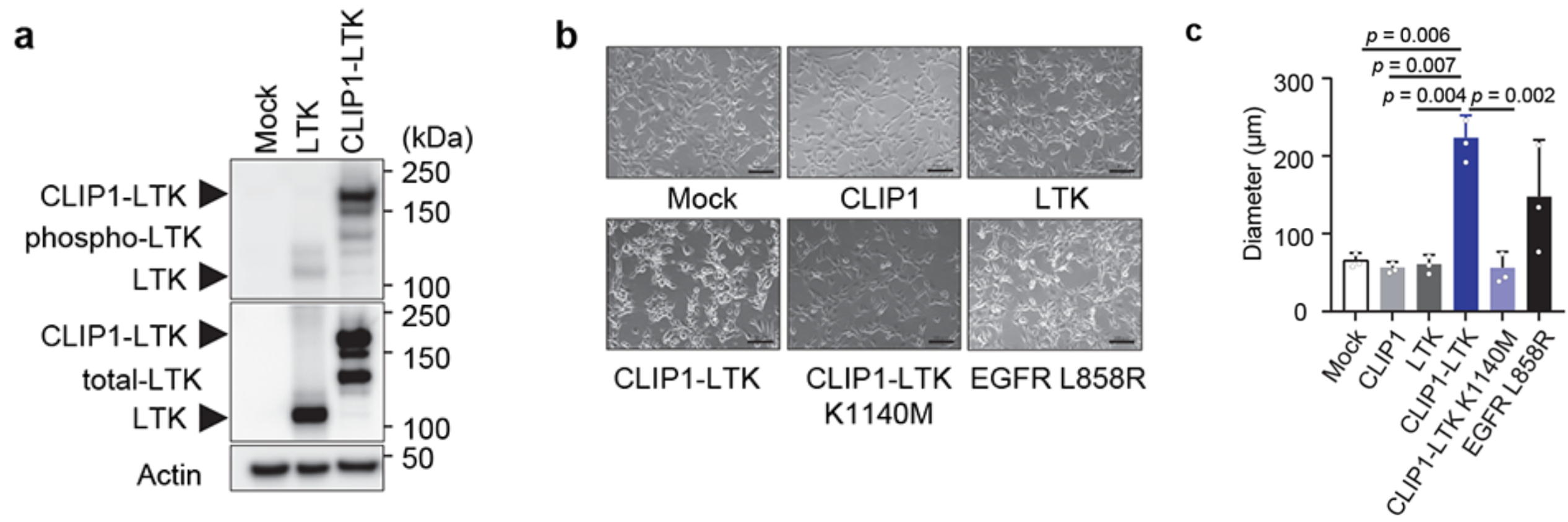
To test whether the CLIP1-LTK fusion protein could activate LTK kinase and drive cellular transformation, NIH3T3 cells were transiently infected with either CLIP1-LTK, wild-type (WT) LTK, or Mock control expression constructs. Western blot analysis detected greater LTK phosphorylation in CLIP1-LTK cells than in WT LTK cells. Both NIH3T3 fibroblasts and Ba/F3 pro-B murine cell lines were transduced with MIGR1 retroviral constructs to analyze transforming potential; cell lines were created to express either human CLIP1, LTK, CLIP1-LTK, CLIP1-LTK-K1140M (a kinase-dead control harboring a K1140M mutation), or Mock control. Again, LTK phosphorylation was observed in CLIP1-LTK cell lines but not in Mock cells, LTK cells, or CLIP1-LTK-K1140M cells, as measured by Western blot. Further, CLIP1-LTK cells showed a round-shape morphology, lack of contact-inhibition, and significantly larger colonies than control. CLIP1-LTK cells even showed larger colonies than positive control cells expressing EGFR-L858R, which has known transforming capacity, although this difference was not statistically significant. The transforming ability of CLIP1-LTK was also confirmed in vivo by subcutaneously injecting the NIH3T3 cell lines into the flank of nude mice. Tumors were only observed in nude mice injected with the CLIP1-LTK cell line. Together, these results suggest that the CLIP1-LTK fusion protein can drive malignant transformation both in vitro and in vivo, dependent on the constitutive activation of LTK kinase.
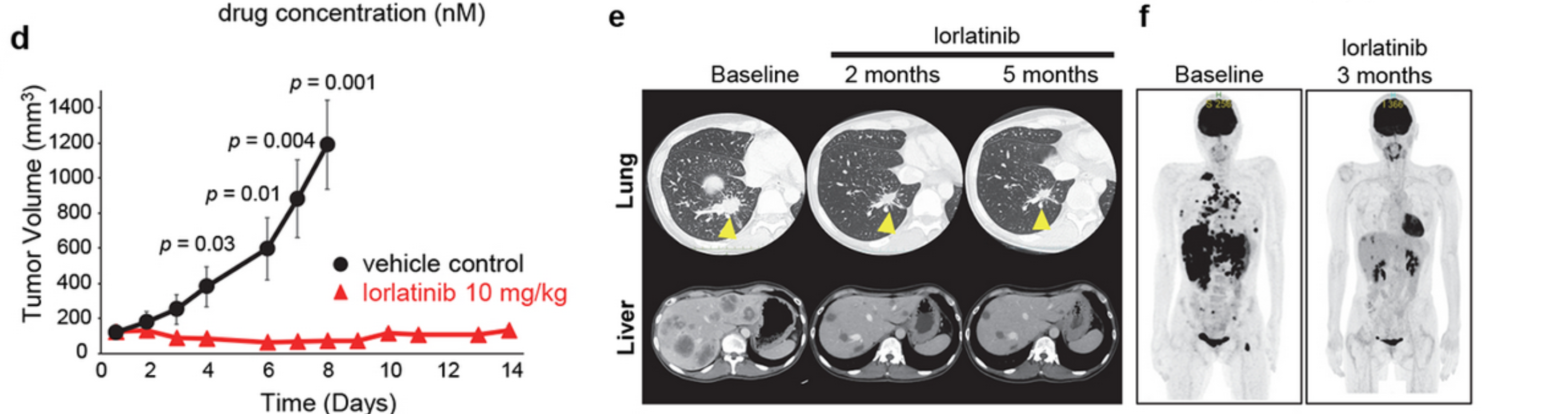
The kinase domain of LTK shares nearly 80% identity to the kinase domain of ALK; given this large degree of homology, the authors hypothesized that ALK tyrosine kinase inhibitors (TKIs) may inhibit the activity of the CLIP1-LTK fusion protein. To test this hypothesis, the NIH3T3-CLIP1-LTK cell lines were treated with varying concentrations of these TKIs. Western blot analysis showed 10nM lorlatinib to be most effective at blocking CLIP1-LTK phosphorylation. In a cell viability assay, the IC50 of lorlatinib was 1.1 nM; the IC50 of other ALK inhibitors were in the 10-20 nM range, and the IC50 of Osimertinib, an EGFR TKI, was >100 nM. Decreased phosphorylation of downstream targets AKT and ERK was observed after a 24-hour treatment with lorlatinib compared to untreated controls; apoptotic markers were also observed. Consistent with these in vitro results, mice that received lorlatinib treatment after having been injected with CLIP1-LTK cells showed significantly inhibited tumor growth compared to controls. The CLIP1-LTK index patient was approved to receive lorlatinib off-label and two- and five-month follow-up CT images show a dramatic clinical response, with shrinkage of both the primary tumor and metastatic tumors. The duration of this response is unknown, and certainly would need to be corroborated by larger clinical studies, yet still warrants an investigation of repurposing available drugs as well as the development of more specific LTK inhibitors. While a larger retrospective screening (n=1,500) for the CLIP1-LTK fusion using the LC-SCRUM-Asia cohort is underway, a preliminary analysis of 542 NSCLC samples identified the alteration in 0.4% (3/542) of samples. Though rare, all 3 patient samples with CLIP1-LTK fusion lacked any other known oncogenic driver, suggesting that CLIP1-LTK is a mutually exclusive driver of lung tumorigenesis. This is also noteworthy in that these patients did not previously have a targeted treatment option available to them. If this cohort is representative of the worldwide population of lung cancer patients, it is predicted that there are more than 7,500 patients with NSCLC who harbor the CLIP1-LTK fusion and may benefit from treatment with lorlatinib, even before the development of selective LTK inhibitors. As NGS panels are developed to detect CLIP1-LTK, FISH can be used to identify patients with this novel oncogenic fusion for treatment.
