Clinical & Research Blog
PROTAC Resistance in Cancer Cells is Mediated by MDR1 Drug Efflux Activity
Selectively inhibiting protein function through proteolysis-targeting chimeras (PROTACs) has emerged as a novel cancer therapeutic strategy. PROTACs are small molecules with two active domains that bind to an E3 ubiquitin ligase and a protein of interest, which results in the ubiquitination and subsequent degradation of the protein of interest. Thus, by hijacking the cell’s ubiquitin-proteasome system, PROTACs can selectively degrade tumor driver proteins and oncogene products. Phase 1/2 clinical trials are currently underway to examine the use of PROTACs in prostate cancer (targeting the androgen receptor) and breast cancer (targeting the estrogen receptor); other recently developed PROTACs target oncogenic kinase proteins, epigenetic proteins, and KRASG12C proteins. Like many cancer therapeutics, cells may exhibit intrinsic or acquired resistance to PROTACs, resulting in diminished therapeutic responses. The goal of this study was to examine the role of the Multidrug Resistance Protein 1 (MDR1), which is encoded by the ABCB1 gene, on PROTAC resistance in several cancer cell lines. MDR1 is a drug efflux protein that has previously been linked to resistance to chemotherapy agents and kinase inhibitors; it has recently been shown to bind to PROTACs. Here, the authors present data to suggest that MDR1-mediated drug efflux contributes to resistance to several PROTAC therapies, but that resistance can be prevented by the coadministration of MDR1 inhibitors.
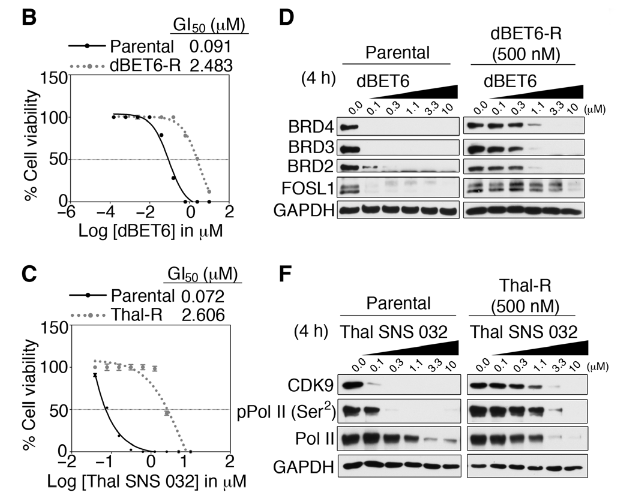
An ovarian cancer cell line (A1847) and a breast cancer cell line (SUM159) were continuously exposed to PROTACs in increasing concentrations; the ovarian cancer cells were exposed to either a bromodomain degrader (dBET6-R) or cyclin-dependent kinase 9 degrader (Thal-R), while the breast cancer cells were exposed to a bromodomain degrader (MZ1-R). The cells that were chronically exposed to PROTACs were more resistant to the effects of these drugs, as demonstrated by a rightward shift in dose-response viability curves compared to parental control cells. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) revealed that treating the chronically exposed cells with increasing PROTAC concentrations was insufficient to degrade the target protein to the extent observed in the parent cells; the drug concentration required to degrade 50% of the target protein (DC50) was much higher for the chronically exposed cells, in some cases even 1000x higher. Thus, these cells had developed PROTAC resistance. To explore the mechanism behind this PROTAC resistance, the authors investigated the top 10 upregulated proteins in the resistant cell lines. They found that MDR1 protein levels were upregulated by 2.5-5x relative to the parental control lines. A rhodamine 123 efflux assay suggested that this increase in MDR1 protein levels also resulted in an increase in efflux activity in these drug-resistant cells, limiting PROTAC accumulation and reducing the degradation of the target protein.
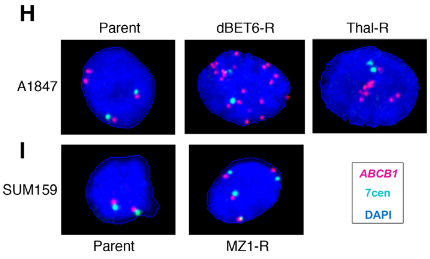
The gene that encodes the MDR1 drug efflux protein, ABCB1, is frequently amplified in chemotherapy-resistant cells, and so fluorescent in situ hybridization (FISH) was used to determine if gene amplification of ABCB1 contributed to the development of PROTAC resistance as well. The drug-resistant ovarian cancer cell line showed significant gene amplification of ABCB1, with a mean of 12 ABC1 copies per nucleus in the Thal-R treatment cells and a mean of 30 ABCB1 copies per nucleus in the dBET6-R treatment cells, compared to only 3-4 centromere control probes per nucleus. In contrast, the drug-resistant breast cancer cells generally had four copies of ABCB1, each signal appearing near a centromere control, which suggests that the copy number increase was due to polysomy rather than amplification in this cell line. As epigenetic mechanisms could also result in an increase in MDR1 expression, the authors examined methylation of the ABCB1 promoter using CpG bisulfate amplicon sequencing. Decreased methylation of the ABCB1 promoter was observed in the ovarian cancer cell line but not in the breast cancer cell line. The authors concluded that ABCB1 amplification and/or decreased methylation of the ABCB1 promoter results in increased MDR1 activity in the PROTAC-resistant ovarian cancer cells but could not draw conclusions about the mechanism of increased MDR1 activity in the PROTAC-resistant breast cancer cells.
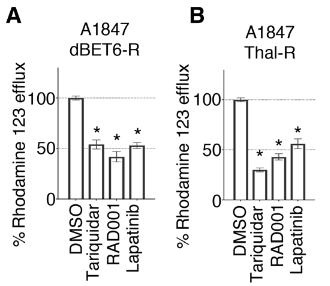
Having linked PROTAC resistance to MDR1 activity, the authors next investigated whether this resistance could be overcome. First, knockdown of ABCB1 reduced cell viability in both the drug-resistant ovarian cancer cell lines and drug-resistant breast cancer cell line, but not in the parental cell line. As the resistant cells were continuously exposed to PROTACs, this result suggests that the ABCB1 knockdown allowed for the accumulation of PROTACs within the cell, restoring the degradation of target proteins and resulting in cell death. Similarly, MDR1 blockade using tariquidar was effective in reducing activity of the MDR1 efflux pump, as measured by the reduced efflux of rhodamine 123; coadministration of tariquidar also increased target protein degradation and inhibited cell viability in the drug-resistant cell lines. However, tariquidar has shown limited clinical success due to toxicities, low drug-drug interactions, and the inability to achieve desired concentrations of tariquidar in tumors; so, the authors next tested the effect of lapatinib, an ErbB receptor inhibitor that is currently FDA-approved for the treatment of HER2-driven cancers that been shown to inhibit MDR1 drug efflux activity, in the PROTAC-resistant cell lines. The lapatinib treatment similarly reduced MDR1 drug efflux activity, resulted in the degradation of the target protein, and caused cell death. Therefore, lapatinib is effective in creating MDR1 blockade to overcome cellular resistance to PROTACs. Clinical trials should explore combining PROTAC treatment with lapatinib for a more durable treatment response.
